Mutation-induced drug resistance DataBase (MdrDB)
About MdrDB
A comprehensive database: The goal of MdrDB is to collate the effects of mutation-induced protein structural changes on binding to small molecules. MdrDB combines protein structures and annotations from the Protein Data Bank (PDB) and Uniprot, with drug data from PubChem and experimentally measured drug effects on wild type proteins and mutants from the Genomics of Drug Sensitivity in Cancer (GDSC) and other databases.
Structure-based: MdrDB provides wild type structures, mutant protein structures, wild type protein-ligand complex structures, and mutant protein-ligand complex structures for protein mutation studies and drug resistance modeling.
Diverse protein mutations: MdrDB contains a variety of mutation types. In addition to single-point substitution mutations, complex mutations such as deletion mutations, insertion mutations, insertion-deletion (indel) mutations, and multi-site mutations are also included.
Current release: MdrDB v.1.0. 2022 contains 100537 samples, generated from 240 proteins (5119 total PDB structures), 2503 mutations, and 440 drugs. 95971 samples are based on available PDB structures, and 4566 samples are based on AlphaFold2 predicted structures.
For more information please see Tutorial.
| Dataset | No. of Samples | Protein Type | Structure-based | Mutation Type | ||
|---|---|---|---|---|---|---|
| Single Substitution | Multiple Substitution | Complex | ||||
| Platinum | 1,008 | Multiple types | × | |||
| TKI | 144 | Kinase | × | × | ||
| RET | 56 | Kinase | × | × | ||
| AIMMS | 311 | Multiple types | × | × | ||
| KinaseMD | 79 | Kinase | × | × | ||
| MdrDB | 100,537 | Multiple types | ||||
-
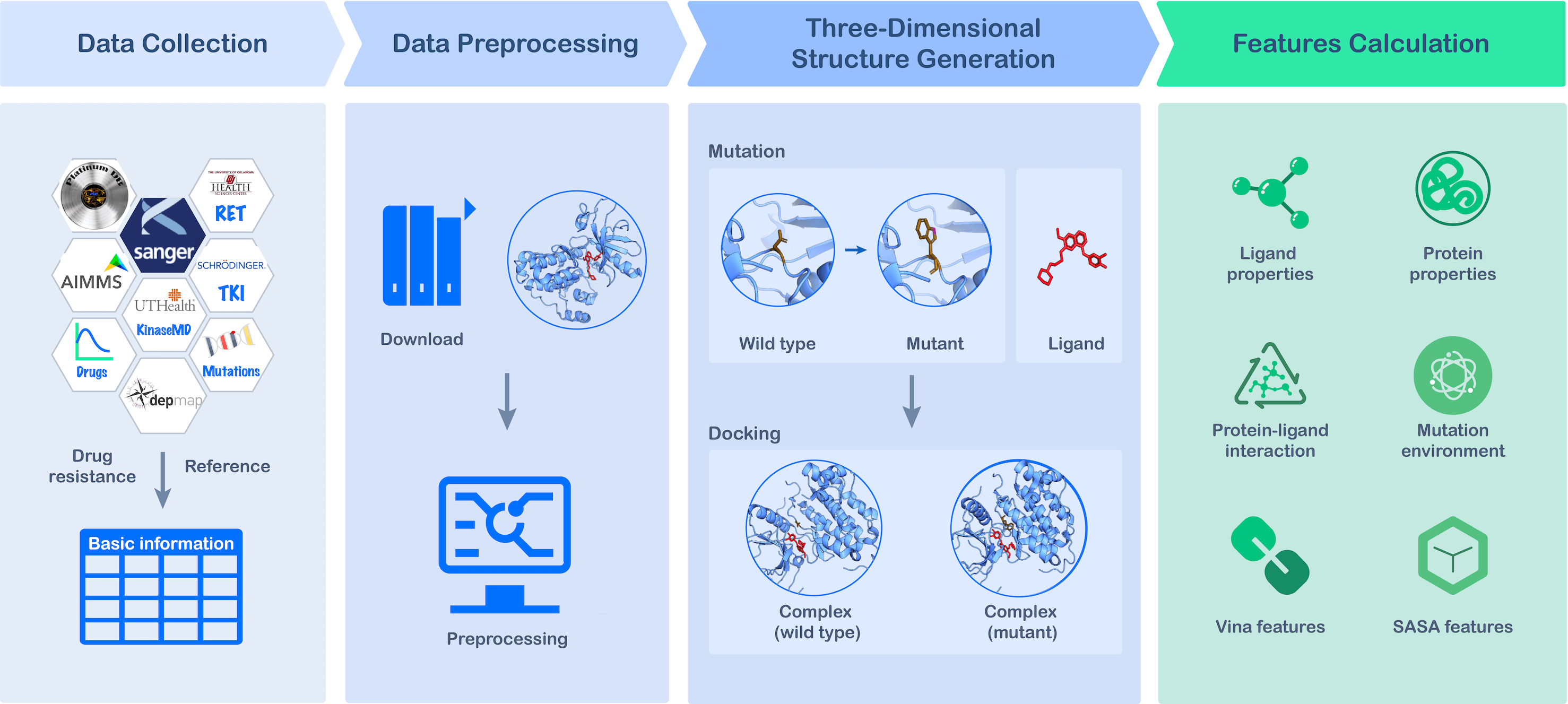
Data Collection
MdrDB contains data from seven publicly available datasets (GDSC, DepMap, AIMMS, KinaseMD, Platinum, TKI, RET) associated with mutation-induced changes in protein-ligand affinity. Binding affinity (ΔΔG), protein information (e.g. Uniprot ID, available PDBs, structural annotations), and drug information (e.g. Generic Name, PubChem CID, SMILES, mechanism annotations) were extracted and integrated with data from other publicly available databases, such as Uniprot, PDB, PubChem, etc.
-
Data Preprocessing
Protein preprocessing: Uniprot ID was first specified for each sample, which is employed to find and download all corresponding available structures (.pdb/.cif) and sequences (.fasta) in PDB. Then the PDB structures were cleaned by removing all water and solvent molecules, as well as binding ligands and extraneous protein chains. Next, an inspection of whether a PDB structure included relevant mutation sites was carried out to exclude non-relevant structures. Finally, protein family, super-family, and domain annotations were queried and documented.
Drug preprocessing: Drug SMILES were first cleaned by removing all salts and neutralizing charges. 3D structures of drugs were then generated based on the resulting SMILES, and polar hydrogens added. Finally, the pharmacological action of the mechanism of drugs was queried and documented.
-
3D Structure Generation
Based on the available wild type structures, mutant protein structures were first generated. For simple single-site substitution mutations, the mutant structures were predicted using the Pymol Mutagenesis Wizard module which accounts for the lowest energy rotamers of the mutated residues. For complex mutations, AlphaFold2 was used to predict mutant structures based on mutated sequences of original structures. For samples without known PDB structures, AlphaFold2 was used to predict both wild type and mutant proteins. Then, drugs were docked to both wild type and mutant proteins to generate protein-ligand complexes.
-
Features Calculation
A total of 146 features were calculated, divided into six types: 18 features that reflect ligand properties, 12 features that represent the wild type and mutant protein differences, 21 features that describe mutation environments, 6 features that model protein-ligand interactions, 59 features of VINA energy functions and 30 features related to solvent accessibility of both ligand and protein.
Statistics
-
100537samples
-
240proteins
-
440drugs
-
2503mutations